







About CSID
What is Congenital Sucrase-Isomaltase Deficiency?
Congenital Sucrase-Isomaltase Deficiency (CSID) is a rare, inherited condition that has several names. It is sometimes referred to as Genetic Sucrase-Isomaltase Deficiency (GSID), Sucrase-Isomaltase Deficiency, Disaccharide Intolerance, or Congenital Sucrose Intolerance.
In This Section
If you think you may have CSID, it is recommended that you schedule a visit to discuss your concerns with a gastroenterologist.
Congenital Sucrase-Isomaltase Deficiency, which can occur in males and females of all ages — infants, children, and adults — reduces the ability to digest (break down to an absorbable form) certain foods one eats that contain sugars or starches. For this reason, enzymes like sucrase-isomaltase are called digestive enzymes.
In people affected by CSID, the enzyme sucrase-isomaltase is either not present at all or has very low levels of enzyme activity in the small intestine. The small intestine is the part of the gastrointestinal (GI) tract that is just beyond the stomach and just before the large intestine. If you have CSID, your body cannot digest complex sugar or starches very well. If you cannot fully digest these foods, you may develop diarrhea, gas, bloating, and abdominal pain after you eat foods that contain sugar (white table sugar) and/or starch (for example, bread, cookies, crackers, and potatoes).1 Undigested foods in your GI tract are eaten by the bacteria that naturally occur in your GI tract, causing these symptoms (Figure 3).
In the 1960s, Weijers and colleagues discovered a GI condition they named CSID. The condition was originally characterized by GI symptoms such as diarrhea, gas and abdominal pain after eating a meal.2
The condition was also associated with the following changes in enzyme activity:2
- Sucrase enzyme activity that was undetectable
- Varying levels of isomaltase enzyme activity
- Maltase enzyme activity that was reduced to nearly one third the normal level

Figure 1. CSID can occur in males and females of all ages — infants, children and adults.
How We Convert Food into Fuel in Our Body
Sucrose (table sugar) is too large to be absorbed from the small intestine. For this reason, sucrose must be digested to its simpler forms, glucose and fructose, before it can be absorbed into the bloodstream and used as an energy source (Figure 2).3 Sucrase is the only naturally-occurring enzyme in humans that can break sucrose down into its simpler forms.
Starches are even larger compounds than sucrose. However, there are several intestinal enzymes that play a role in the digestion of the starches we eat, including:
- Sucrase-isomaltase
- Maltase-glucoamylase
- α-amylases
So, if you have CSID and the enzyme sucrase-isomaltase is missing from your small intestine or isn’t working or isn’t working well, you may or may not have GI symptoms after eating foods that contain starches. While there is only one naturally-occurring enzyme (sucrase) in our bodies that digests sucrose, there are multiple enzymes in our bodies that act to digest starches. Because only sucrase can digest sucrose, eating foods that contain sucrose usually causes worse GI symptoms in patients with CSID than eating foods that contain starches because some of those starches may be broken down in part by one of the two enzymes other than sucrase-isomaltase.
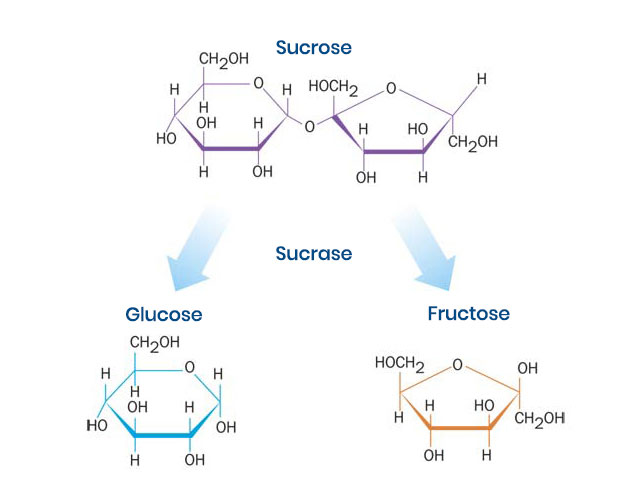
Figure 2. Sucrose cannot be absorbed as is and is useless as an energy source unless it is broken down into its simpler components, glucose and fructose.
What are the Consequences of Not Absorbing Sugars and Starches?
Common GI symptoms associated with CSID that follow a meal containing sucrose or starch include watery diarrhea, nausea, bloating and gassiness, abdominal distention (swelling), and pain.4
Failure to break down and absorb foods containing sucrose and starch from the small intestine can:4
- Slow the normal flow of foods you’ve eaten from the stomach into the small intestine
- Accelerate the movement of foods from the small intestine into the large intestine
- Contribute to a reduced absorption of other dietary nutrients, such as other starches and sugars, fats, and proteins
- Interfere with hormonal regulations of your GI function
- Lead to GI symptoms
- Lead to low body weight or malnutrition
For this reason, individuals with CSID are at risk for chronic malnutrition. Indeed, failure to thrive is one of the primary characteristics of the disease in children.5
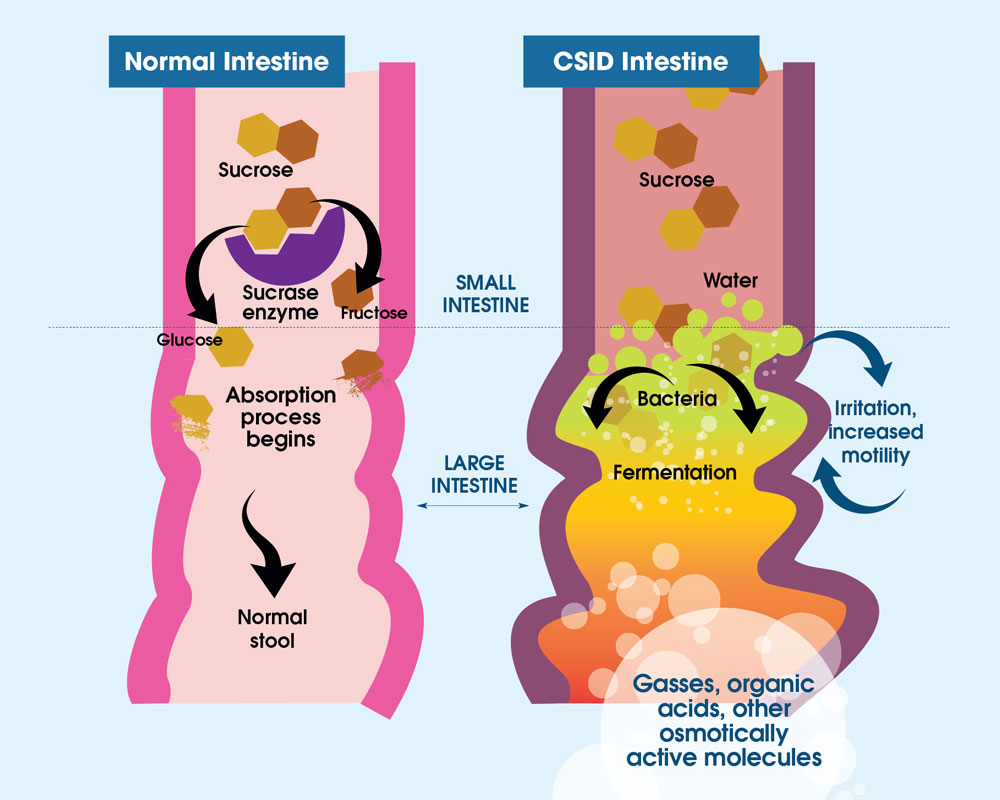
Figure 3. Normally, sucrose is digested and metabolized (broken down) by sucrase (purple object) into two subunits, glucose (gold object) and fructose (brown object). These smaller sugars are able to be absorbed by the small intestine. With CSID, sucrase is absent or not working well and sucrose is not broken down into its two subunits, glucose and fructose. As sucrose is too large to be absorbed by the small intestine, it becomes food for the bacteria, called microflora, that normally live in the large intestine. The way this microflora breaks down sugars is by fermentation, and the byproducts of fermentation are gases, organic acids, or other molecules. The presence of these byproducts causes abdominal distention (swelling) and pain, and the retention of water in the intestine, which produces watery diarrhea.
References
- Treem WR. Clinical heterogeneity in Congenital Sucrase-Isomaltase Deficiency. J Pediatr. 1996;128(6):727-729. doi:10.1016/s0022-3476(96)70320-7
- Weijers HA, van de Kamer JH, Mossel DA, Dicke WK. Diarrhoea caused by deficiency of sugar-splitting enzymes. Lancet. 1960;2(7145):296-297. doi:10.1016/s0140-6736(60)91381-7
- Gray GM. Carbohydrate digestion and absorption. Role of the small intestine. N Engl J Med. 1975;292(23):1225-1230. doi:10.1056/NEJM197506052922308
- Treem WR. Congenital Sucrase-Isomaltase Deficiency. J Pediatr Gastroenterol Nutr. 1995;21(1):1-14. doi:10.1097/00005176-199507000-00001
- Antonowicz I, Lloyd-Still JD, Khaw KT, Shwachman H. Congenital Sucrase-Isomaltase Deficiency. Observations over a period of 6 years. Pediatrics. 1972;49(6):847-853.