






About CSID
What is Congenital Sucrase-Isomaltase Deficiency?
Congenital Sucrase-Isomaltase Deficiency (CSID) is a rare genetic disorder, first discovered in 1960 by Weijers and colleagues,1 caused by pathogenic mutations in the sucrase-isomaltase gene (SI).
In This Section
It is a disease of the small intestine characterized primarily by a failure to metabolize sucrose for intestinal absorption. The disease was originally clinically defined by undetectable sucrase activity (enzyme responsible for sucrose metabolism), a decrease in maltase activity (one of the enzymes involved in the metabolism of the remnant starch sugar, maltose) to nearly one third the normal level, and varying degrees of isomaltase activity (one of the enzymes responsible for the metabolism of complex, branched starch bonds).1
Animation: Sucrase Hydrolysis of Sucrose © The McGraw-Hill Companies, Inc.
This disease has historically been described primarily as an autosomal recessive disorder associated with homozygous or compound heterozygous genotypes of pathogenic SI gene mutations. So far, 37 distinct SI gene mutations have been identified in patients with a confirmed diagnosis of CSID based on enzyme activity from a small bowel biopsy.3-11
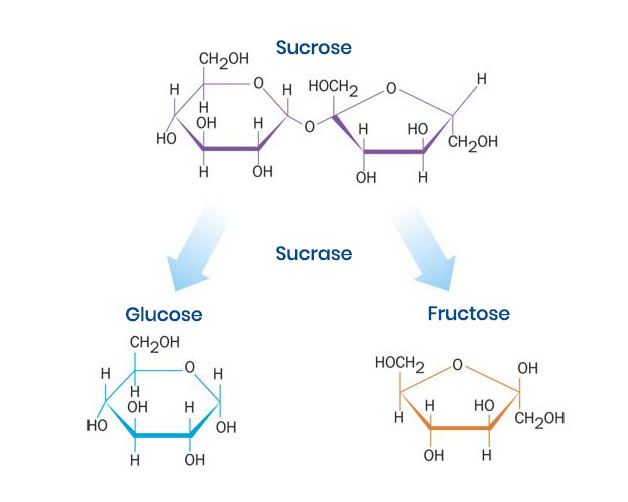
Figure 1. Sucrose metabolism by sucrase. Sucrase metabolizes sucrose into its constituent subunits, glucose and fructose, to increase their gastrointestinal absorption.2
The SI gene is very large (more than 100 kilobase pairs), and there are more than 2,000 known, rare mutations, many of which have not been thoroughly investigated.12 Recent research has found that CSID patients have a spectrum of phenotypic expression and severity associated with remnant enzyme activity, total carbohydrate consumption, and a variety of genotypes including SI homozygotes, simple heterozygotes, and compound heterozygotes.13 In addition, heterozygotes with an SI mutation associated with CSID have been shown to have an increased risk of IBS.12,14 This range of inheritance patterns and broad phenotypic expression suggests CSID may be more prevalent than originally thought.15
Most importantly, the pathophysiology of CSID is defined by a lack of functional sucrase activity within the small intestine, the enzyme that metabolizes sucrose to its constituent glucose and fructose subunits (Figure 1). While sucrose intolerance is a cardinal feature, CSID can manifest with varying degrees of starch intolerance. Symptom severity is a function of residual sucrase and isomaltase activity, the amount of sugar and starch consumed, the presence of other foods that may have a buffering effect, and the rate of gastric transit.15
Sucrase-isomaltase deficiency has clinically significant consequences, as failure to absorb dietary sucrose and starch may impact the absorption of other nutrients, as well as the hormonal regulation of gastrointestinal function. This is because unabsorbed carbohydrates can accelerate small intestine transit and contribute to malabsorption of starch, fat, and monosaccharides.4 Normal postprandial increases in hormones such as insulin, C-peptide, and gastric inhibitory peptide may be disrupted.15 For these reasons, patients with CSID are at risk for chronic malnutrition. Indeed, failure to thrive or low body mass index are characteristics of the disease.16
What is Sucrase-Isomaltase?
Sucrase-isomaltase is an integral glycoprotein heterodimer enzyme involved in carbohydrate metabolism and expressed in the brush border membrane of cells lining the small intestine.15 Sucrase-isomaltase is synthesized, glycosylated, and transported to the apical membrane of the microvilli in the intestinal lumen. In the intestinal brush border, the two enzyme subunits dissociate and the two individual subunits become membrane bound and are associated by noncovalent, strong ionic interactions.15,17-19
What Are the Presenting Symptoms of CSID?
The primary symptom of CSID is mild to severe, chronic, watery diarrhea.15 In addition, fermentation of excess, unabsorbed dietary saccharides by resident intestinal colonic bacteria can result in gassiness, abdominal distention, pain, accelerated motility, and explosive, acidic diarrhea.15 Chronic, watery diarrhea and failure to thrive are the most common symptoms of CSID in infants and toddlers.15,16 Other manifestations include colic, irritability, excoriated buttocks, diaper rash, and vomiting.15,16,20 However, symptoms of CSID do not manifest in infants until they begin to ingest sucrose and starch-containing foods (for example, juices, most common baby foods, medications sweetened with sucrose), because breast milk does not contain sucrose.15 A small number of patients may need to be hospitalized for diarrhea-induced dehydration, malnutrition, muscle wasting, and weakness.20 In some ethnic populations that maintain a traditional protein-rich diet, notably Greenland Eskimos and some Alaskan and Canadian Natives, a low-carbohydrate, high-protein, high-fat diet may mask symptoms.15,20
Because it is genetic, CSID is not a disease that a patient can outgrow. Indeed, symptoms persist in adults, but may appear less severe than those experienced by children due to diet and the length of the GI tract. However, in patients with CSID with some residual SI activity, expression of the SI enzyme in the brush border may be induced by hormonal factors, such as corticosteroids and thyroxine, and dietary factors, such as a high-sucrose, high carbohydrate diet.15
As with pediatric patients, the clinical presentation of CSID in adults varies. In some adults, the symptoms may be limited to an increase in bowel movement (BM) frequency, reduced stool consistency (looser stools or watery stools), abdominal distention, and flatulence (gas). Episodic watery diarrhea may also occur after eating a meal that contains high levels of sucrose. In some individuals affected with CSID, diarrhea may alternate with constipation, particularly when taking common antidiarrheal medications, which may lead to a misdiagnosis of another gastrointestinal (GI) condition, such as irritable bowel syndrome-alternating (IBS-A).15
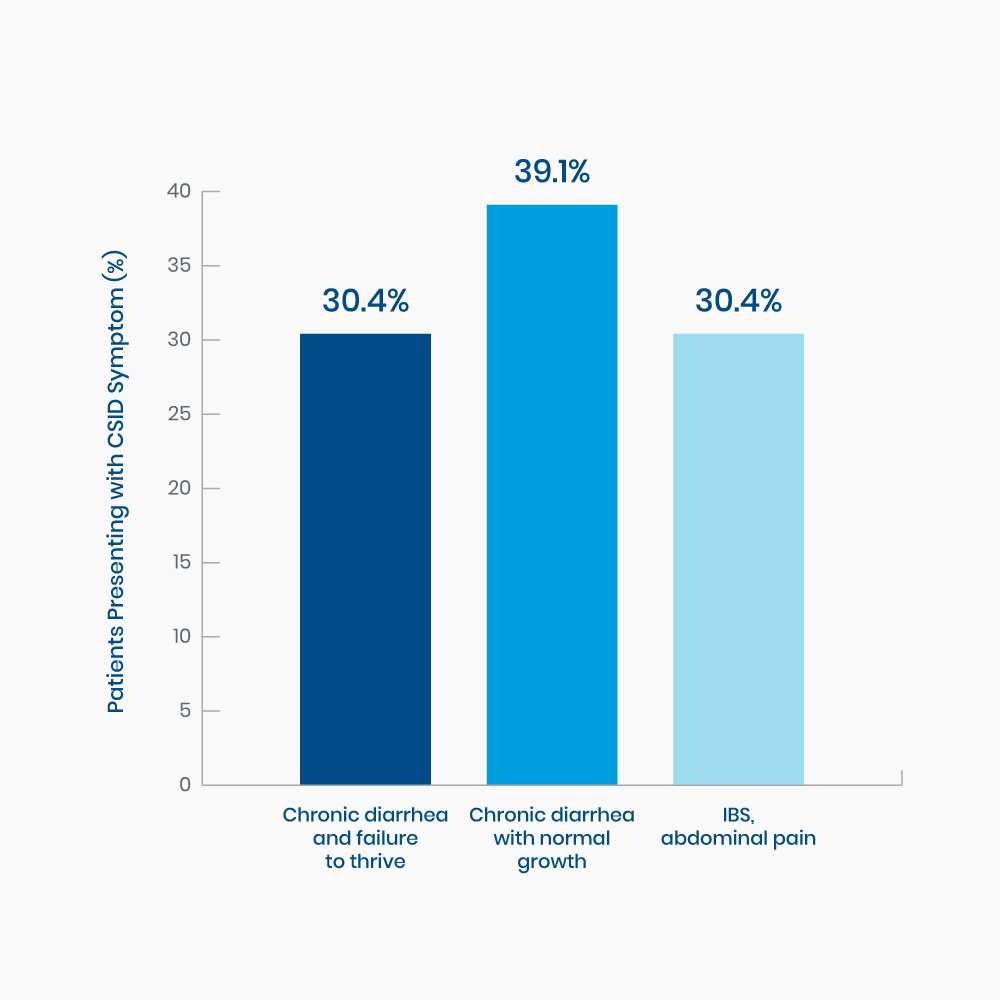
Figure 2. Range of presenting symptoms in 23 pediatric patients diagnosed with CSID15
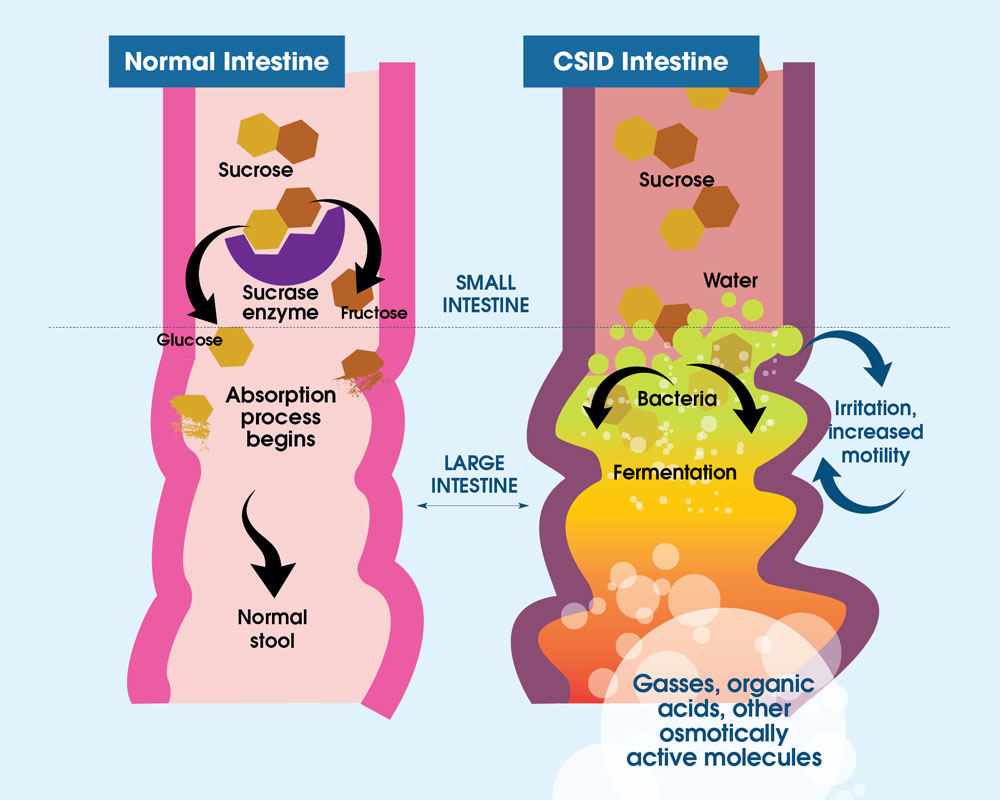
Figure 3. In individuals with normal digestion of sucrose, sucrose is metabolized by sucrase (purple) into two subunits, glucose (gold) and fructose (brown). With CSID, sucrase is absent or dysfunctional, and sucrose cannot be metabolized. Thus, sucrose is not absorbed by the small intestine and undergoes bacterial fermentation in the intestine. The fermentation byproducts are gases, organic acids, or other osmotically-active molecules, causing abdominal distention and pain, and water retention producing watery diarrhea.
References
- Weijers HA, van de Kamer JH, Mossel DA, Dicke WK. Diarrhoea caused by deficiency of sugar-splitting enzymes. Lancet. 1960;2(7145):296-297. doi:10.1016/s0140-6736(60)91381-7
- Gray GM. Carbohydrate digestion and absorption. Role of the small intestine. N Engl J Med. 1975;292(23):1225-1230. doi:10.1056/NEJM197506052922308
- Alfalah M, Keiser M, Leeb T, et al. Compound heterozygous mutations affect protein folding and function in patients with Congenital Sucrase-Isomaltase Deficiency. Gastroenterology. 2009;136(3):883-92. doi:10.1053/j.gastro.2008.11.038
- Gericke B, Amiri M, Naim HY. The multiple roles of sucrase-isomaltase in the intestinal physiology. Mol Cell Pediatr. 2016;3(1):2. doi: 10.1186/s40348-016-0033-y
- Jacob R, Zimmer KP, Schmitz J, et al. Congenital Sucrase-Isomaltase Deficiency arising from cleavage and secretion of a mutant form of the enzyme. J Clin Invest. 2000;106(2):281-7. doi:10.1172/JCI9677
- Keiser M, Alfalah M, Pröpsting MJ, et al. Altered folding, turnover, and polarized sorting act in concert to define a novel pathomechanism of Congenital Sucrase-Isomaltase Deficiency. J Biol Chem. 2006;281(20):14393-9. doi:10.1074/jbc.M513631200
- Naim HY, Heine M, Zimmer KP. Congenital Sucrase-Isomaltase Deficiency: heterogeneity of inheritance, trafficking, and function of an intestinal enzyme complex. J Pediatr Gastroenterol Nutr. 2012;55(suppl 2):S13-20. doi: 10.1097/01.mpg.0000421402.57633.4b
- Ritz V, Alfalah M, Zimmer KP, et al. Congenital Sucrase-Isomaltase Deficiency because of an accumulation of the mutant enzyme in the endoplasmic reticulum. Gastroenterology. 2003;125(6):1678-85. doi:10.1053/j.gastro.2003.09.022
- Sander P, Alfalah M, Keiser M, et al. Novel mutations in the human sucrase-isomaltase gene (SI) that cause congenital carbohydrate malabsorption. Human Mutat. 2006;27(1):119. doi: 10.1002/humu.9392
- Spodsberg N, Jacob R, Alfalah M, et al. Molecular basis of aberrant apical protein transport in an intestinal enzyme disorder. J Biol Chem. 2001;276(26):23506-10. doi:10.1074/jbc.C100219200
- Uhrich S, Wu Z, Huang J, Scott CR. Four mutations in the SI gene are responsible for the majority of clinical symptoms of CSID. J Pediatr Gastroenterol Nutr. 2012; 55(2):S34-5. doi:10.1097/01.mpg.0000421408.65257.b5
- Garcia-Etxebarria K, Zheng T, Bonfiglio F, et al. Increased prevalence of rare sucrase-isomaltase (SI) pathogenic variants in irritable bowel syndrome patients. Clin Gastroenterol Hepatol. 2018;16(10):1673-76. doi:10.1016/j.cgh.2018.01.047
- Cohen SA. The clinical consequences of sucrase-isomaltase deficiency. Mol Cell Pediatr. 2016;3:5. doi: 10.1186/s40348-015-0028-0
- Henström M, Diekmann L, Bonfiglio F, et al. Functional variants in the sucrase-isomaltase gene associate with increased risk of irritable bowel syndrome. Gut. 2018;67(2):263-270. doi:10.1136/gutjnl-2016-312456
- Treem WR. Congenital Sucrase-Isomaltase Deficiency. J Pediatr Gastroenterol Nutr. 1995;21(1):1-14. doi:10.1097/00005176-199507000-00001
- Antonowicz I, Lloyd-Still JD, Khaw KT, Shwachman H. Congenital Sucrase-Isomaltase Deficiency. Observations over a period of 6 years. Pediatrics. 1972;49(6):847-853.
- Naim HY, Sterchi EE, Lentze MJ. Biosynthesis of human sucrase-isomaltase complex. Differential O-glycosylation of the sucrase subunit correlates with its position with the enzyme complex. J Biol Chem. 1988;263(15):7242-7253.
- Naim HY, Roth J, Sterchi EE, et al. Sucrase-isomaltase deficiency in humans. Different mutations disrupt intracellular transport, processing, and function of an intestinal brush border enzyme. J Clin Invest. 1988;82(2):667-679. doi:10.1172/JCI113646
- Hauri HP, Quaroni A, Isselbacher KJ. Biogenesis of intestinal plasma membrane: posttranslational route and cleavage of sucrase-isomaltase. Proc Natl Acad Sci USA. 1979;76(10):5183-5186. doi:10.1073/pnas.76.10.5183
- Gudmand-Høyer E. Sucrose malabsorption in children: a report of thirty-one Greenlanders. J Pediatr Gastroenterol Nutr. 1985;4(6):873-877. doi:10.1097/00005176-198512000-00005